Introduction
Overview
Teaching: 0 min
Exercises: 0 minQuestions
Introducción al taller
Objectives
Conocer los objetivos del taller
En este curso aprenderemos a trabajar con datos de secuenciación masiva, y en particular aprenderemos a analizar datos de RNAseq. Sin embargo, para observar como difieren distintos tipos de datos de secuenciación masiva, también analizaremos ligeramente datos de DNAseq y ATACseq.
Para realizar este curso es necesario haber descomprimido el archivo Clase_Bioinformatica.zip (enviado por el instructor) en una carpeta accesible para el sistema de Unix que este utilizando (Ver lesson-setup)
Key Points
¿Que tipo de datos utilizaremos?
Ordenando los datos de secuenciacion masiva
Overview
Teaching: 60 min
Exercises: 30 minQuestions
¿Como proceso los archivos obtenidos de secuenciación masiva?
Objectives
Aprender a renombrar y mover archivos usando funciones de bash
Explorar y ordenar los archivos del curso
Primero creamos la carpeta ubuntu para descomprir en esa carpeta el archivo Taller_NGS.zip
cd /mnt/c/
mkdir ubuntu/
cd ubuntu
pwd
/mnt/c/ubuntu/Taller_NGS
Para ver si hemos descomprimido bien todos los archivos revisamos con ls. Añadimos la opción -R para visualizar también el contenido incluido en la carpeta reference_genome
ls -1R
.:
ERR023691_1.fastq.gz
ERR023691_2.fastq.gz
metadata.txt
reference_genome
SRR13826529_1.fastq.gz
SRR13826529_2.fastq.gz
SRR13826530_1.fastq.gz
SRR13826530_2.fastq.gz
SRR13826531_1.fastq.gz
SRR13826531_2.fastq.gz
SRR13826532_1.fastq.gz
SRR13826532_2.fastq.gz
SRR13826533_1.fastq.gz
SRR13826533_2.fastq.gz
SRR13826534_1.fastq.gz
SRR13826534_2.fastq.gz
SRR13826535_1.fastq.gz
SRR13826535_2.fastq.gz
./reference_genome:
Saccharomyces_cerevisiae.R64-1-1.57.gff3
Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa
El archivo metadata.txt contiene información sobre el origen de cada archivo de secuenciación, indicando la cepa de levadura, la réplica biológica, el experimento (HIGH NITROGEN,LOW NITROGEN,YPD), y el tipo de ensayo que se obtuvo de ese experimento (RNAseq, DNAseq, ATACseq).
Para revisarlo en pantalla (y aprovechando que es un archivo tabulado relativamente pequeño) usaremos cat
cat metadata.txt
WExNA 1 RNAseq HIGH_N SRR13826535_1.fastq.gz SRR13826535_2.fastq.gz
WExNA 2 RNAseq HIGH_N SRR13826534_1.fastq.gz SRR13826534_2.fastq.gz
WExNA 3 RNAseq HIGH_N SRR13826533_1.fastq.gz SRR13826533_2.fastq.gz
WExNA 1 RNAseq LOW_N SRR13826532_1.fastq.gz SRR13826532_2.fastq.gz
WExNA 2 RNAseq LOW_N SRR13826531_1.fastq.gz SRR13826531_2.fastq.gz
WExNA 3 RNAseq LOW_N SRR13826530_1.fastq.gz SRR13826530_2.fastq.gz
WE 1 DNAseq YPD ERR023691_1.fastq.gz ERR023691_2.fastq.gz
WExNA 1 ATACseq HIGH_N SRR13826529_1.fastq.gz SRR13826529_2.fastq.gz
Aprovecharemos los datos de metadata para renombrar los archivos de secuenciación. Para lograr esto de forma automática correremos un loop que renombrará cada archivo registrado en metadata (columnas $5 y $6) con el nombre cepa ($1), replicado ($2), tipo de ensayo($3), y experimento ($4), a lo que le incluiremos la extension “.fq.gz”. Este script lo correremos dos veces, uno para cada archivo de las reads pareadas (“_1” o “_2”)
cat metadata.txt | while read line;do name1=$(echo "$line"|awk '{print $5}');name2=$(echo "$line"|awk '{print $1"_"$2"_"$3"_"$4}');mv "$name1" "${name2}_1.fq.gz";done
cat metadata.txt | while read line;do name1=$(echo "$line"|awk '{print $6}');name2=$(echo "$line"|awk '{print $1"_"$2"_"$3"_"$4}');mv "$name1" "${name2}_2.fq.gz";done
Cuidado!
Para que este script sea exitoso, todos los archivos deben estar en la carpeta del script, y no deberian haber espacios en ninguno de los nombres a utilizar (lo que en general es buena práctica cuando se trabaja en unix). Como funciona este script? Este script es un poco complejo, combina los programas CAT, AWK, y MV, y además incluye un pipe (“|”) y un loop “WHILE READ”. El loop “while read” nos permite generar una variable (“line”) que será una fila entera del archivo metadata.txt (que lo alimentamos con cat y el pipe |) El hecho de que sea un loop significa que hará su tarea fila por fila hasta que llegue al final del archivo metadata.txt. Lo que hay entre “do” y “done” son los comandos necesarios para encontrar los archivos originales y renombrarlos, siempre usando la información obtenida por el “while read” desde metadata.txt
Revisemos los archivos renombrados
ls
WE_1_DNAseq_YPD_1.fq.gz
WE_1_DNAseq_YPD_2.fq.gz
WExNA_1_ATACseq_MS300_1.fq.gz
WExNA_1_ATACseq_MS300_2.fq.gz
WExNA_1_RNAseq_MS300_1.fq.gz
WExNA_1_RNAseq_MS300_2.fq.gz
WExNA_1_RNAseq_MS60_1.fq.gz
WExNA_1_RNAseq_MS60_2.fq.gz
WExNA_2_RNAseq_MS300_1.fq.gz
WExNA_2_RNAseq_MS300_2.fq.gz
WExNA_2_RNAseq_MS60_1.fq.gz
WExNA_2_RNAseq_MS60_2.fq.gz
WExNA_3_RNAseq_MS300_1.fq.gz
WExNA_3_RNAseq_MS300_2.fq.gz
WExNA_3_RNAseq_MS60_1.fq.gz
WExNA_3_RNAseq_MS60_2.fq.gz
metadata.txt
reference_genome
Ahora es buen momento para ordenar nuestros archivos en distintas carpetas, una para cada tipo de ensayo. Para hacerlo de forma automatica en bash usaremos en conjunto las herramientas “find” y “mv”
Primero tendremos que crear los directorios
mkdir RNAseq
mkdir ATACseq
mkdir DNAseq
El comando para mover los archivos a sus respectivas carpetas (aprovechando que incluimos el tipo de ensayo al renombrar los archivos) es el siguiente
find *_RNAseq* -exec mv -t RNAseq/ {} +
find *_ATACseq* -exec mv -t ATACseq/ {} +
find *_DNAseq* -exec mv -t DNAseq/ {} +
Key Points
Usar archivos de textos como metadata para ordenar experimentos bioinformaticos
Análisis de calidad
Overview
Teaching: 60 min
Exercises: 30 minQuestions
¿Cuál es la calidad de mis datos de secuenciación?
Objectives
Analizar la calidad de las lecturas provenientes de secuenciacion masiva usando FASTQC y MULTIQC
El formato fastq
Todas las reads que obtuvimos de los distintos ensayos tienen una extension tipo fastq (*.fq o *.fastq), que en la práctica son simplemente archivos de texto con la secuencias nucleotidicas y la calidad de la secuenciación de estas. En nuestro caso, las reads estan comprimidas, por lo tanto además tienen la extension .gz Podemos mirar las primeras lineas de estos archivos fastq primero descomprimiendo (usando gzip) y luego usando el comando “head” que imprimirá solo las primeras lineas en pantalla (estos archivos generalmente tienen miles de lineas!)
gzip -k -d DNAseq/WE_1_DNAseq_YPD_1.fq.gz
head -n 4 DNAseq/WE_1_DNAseq_YPD_1.fq
@IL29_4505:7:1:1072:8738#5/1
TTGCCTTTTTTTGTTGACAATAAATTCCCTTCTGAAGCCACCGGGGACATCTACACCACTTGCTGATGACACTTTAGTATGGTGCTCACTTTCAGGTGTTACATTCTC
+
BABABBAAAABBBBBB:BBAB>,>BBBBBBBB>>86A?A=BBBBAB-B??,7237BB:BBB:A>@.=?.A7,743?),940527?B>@=B<A+A6;>B>><B?;>+,?
Entonces head -n 4 nos muestra las primeras cuatro lineas del archivo WE_1_DNAseq_YPD_1.fq. Estas primeras cuatro lineas corresponden a la información completa de la primera read (si hacen head -n 8 verán la información de las primeras dos reads) La primera linea es el nombre que se le asigna a la read (que comienza con una @), donde por ejemplo se indica el modelo del secuenciador (IL29_4505), y al final a cual de los dos pares corresponde (/1) (en caso de reads pareadas)
@IL29_4505:7:1:1072:8738#5/1
En la segunda linea esta la secuencia nucleotidica obtenida
TTGCCTTTTTTTGTTGACAATAAATTCCCTTCTGAAGCCACCGGGGACATCTACACCACTTGCTGATGACACTTTAGTATGGTGCTCACTTTCAGGTGTTACATTCTC
En la cuarta linea esta la calidad de la secuenciacion para cada base de la secuencia obteniva
BABABBAAAABBBBBB:BBAB>,>BBBBBBBB>>86A?A=BBBBAB-B??,7237BB:BBB:A>@.=?.A7,743?),940527?B>@=B<A+A6;>B>><B?;>+,?
Cada letra,número, o simbolo representa un valor de calidad de 1 a 40:
Quality encoding: !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHI
| | | | |
Quality score: 0........10........20........30........40
Estos valores (Phred score) representan para cada base nucleotidica la probabilidad de que hayan sido designadas correctamente. Este valor es en base logaritmica, por lo tanto un valor Phred de 10 refiere a una confianza del 90% de que la base haya sido asignada correctamente.
PhredQuality Score Probability of incorrect base call Base call accuracy
10 1 in 10 90%
20 1 in 100 99%
30 1 in 1000 99.9%
40 1 in 10,000 99.99%
FASTQC
El programa FASTQC nos ayudará visualizar el phred score de cada base secuenciada de todas nuestras reads, además de otros parametros.
Primero analizaremos el archivo WE_1_DNAseq_YPD_1.fq.gz (notar que no es necesario descomprimir el archivo para que lo ocupe FASTQC)
fastqc DNAseq/WE_1_DNAseq_YPD_1.fq.gz
FASTQC genera un informe en formato HTML (más un archivo zip) del análisis de este archivo de secuencia (por defecto deja los informes en la carpeta del archivo analizado). Podemos abrir este informe usando un explorador web.
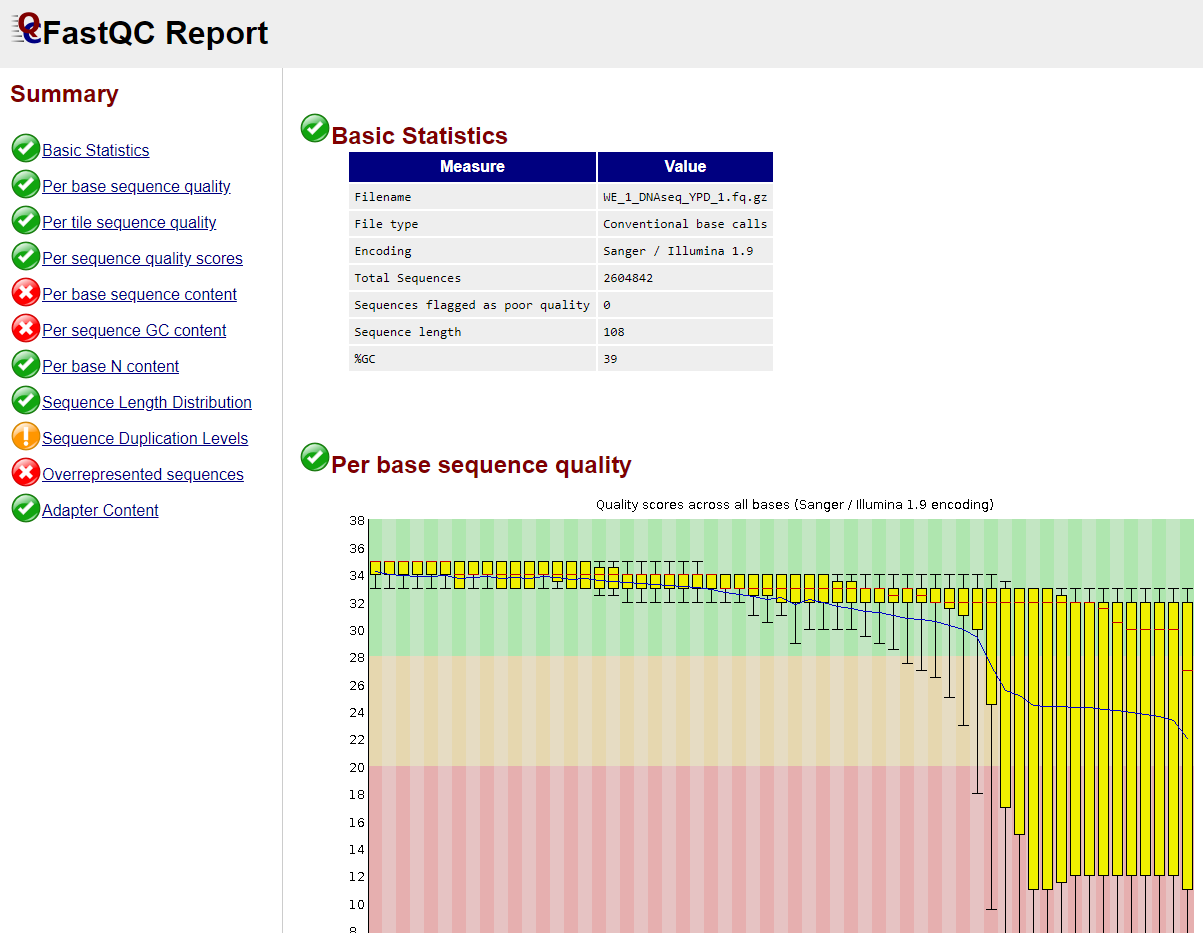
Para mayor información sobre el informe de FASTQC pueden visitar el siguiente link: https://hbctraining.github.io/Intro-to-rnaseq-hpc-salmon/lessons/qc_fastqc_assessment.html
Ahora ejecutemos FASTQC para todos nuestros archivos. Para que FASTQC trabaje más rápido, podemos aumentar el número de CPUs que utilizará con la opción -t (el máximo depende del computador que estemos usando, pueden revisar el número de CPUs que tiene con el comando “htop”)
Primero crearemos carpetas donde guardaremos los informes de estas reads crudas (raw)
mkdir DNAseq/fastqc_raw
mkdir RNAseq/fastqc_raw
mkdir ATACseq/fastqc_raw
fastqc -t 8 -o DNAseq/fastqc_raw DNAseq/*.fq.gz
fastqc -t 8 -o ATACseq/fastqc_raw ATACseq/*.fq.gz
fastqc -t 8 -o RNAseq/fastqc_raw RNAseq/*.fq.gz
En total se generaron 16 archivos HTML, y no es muy conveniente revisarlos uno a uno. El programa MULTIQC nos ayudará a generar un informe de los informes de FASTQC (Este programa también sirve para compilar otros tipos de informes de una variedad aplicaciones bioinformaticas)
multiqc -o DNAseq DNAseq/fastqc_raw
multiqc -o ATACseq ATACseq/fastqc_raw
multiqc -o RNAseq RNAseq/fastqc_raw
El output de multiqc es también un archivo HTML (que especificamos que se guardara en la carpeta que analizamos). Gracias a multiqc podemos revisar los 12 archivos de secuenciación de RNAseq en el mismo gráfico:
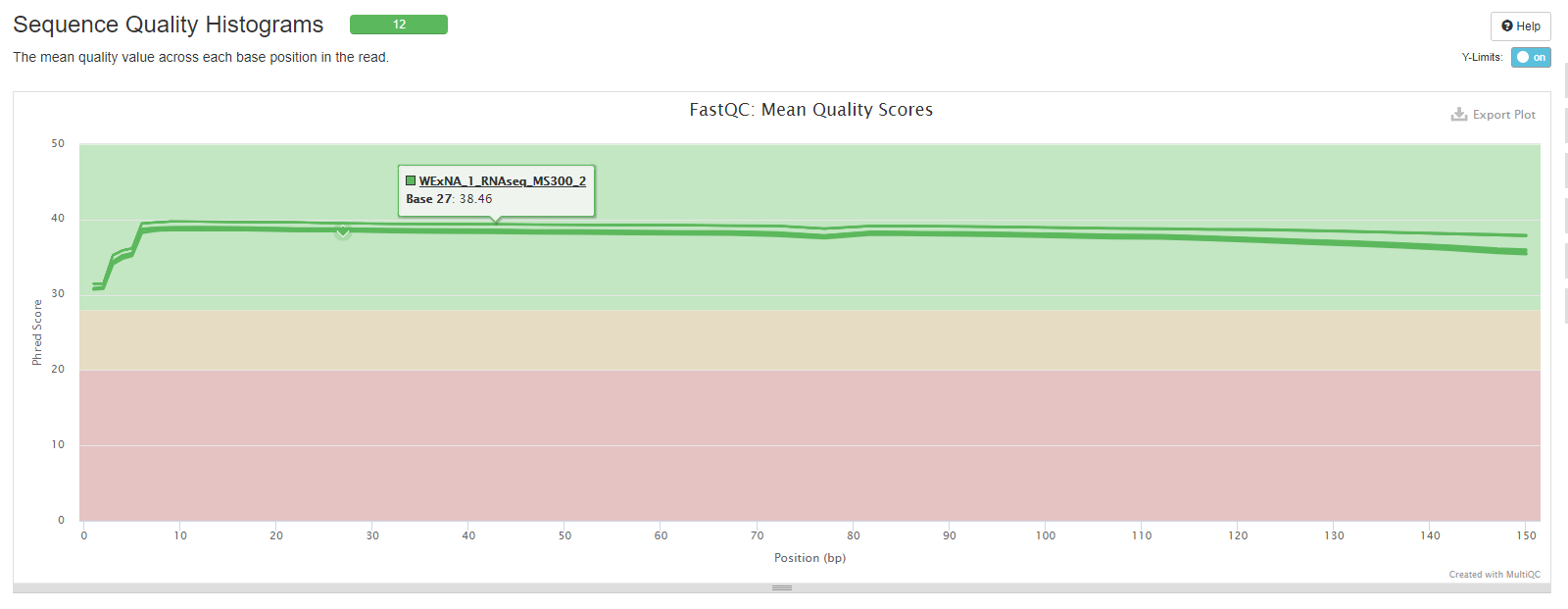
Ahora sabemos que nuestras reads de RNAseq y ATACseq en general tienen buena calidad, sin embargo las reads de DNAseq presentan varios problemas (baja calidad en el extremo 3’, presencia de adaptadores)
Procesamiento de reads con FASTP
Para arreglar nuestras reads, por ejemplo cortando los segmentos de baja calidad y eliminando adaptadores, utilizaremos el programa FASTP Algo muy importante de FASTP es que acepta como input archivos de reads pareadas, por lo que si un miembro del par de las reads tiene mala calidad, fastp podrá eliminar ambas reads (que estan seperadas en archivos _1 y _2) El siguiente comando limpiará las reads de DNAseq:
fastp --in1 DNAseq/WE_1_DNAseq_YPD_1.fq.gz --in2 DNAseq/WE_1_DNAseq_YPD_2.fq.gz --out1 DNAseq/cleaned_WE_1_DNAseq_YPD_1.fq.gz --out2 DNAseq/cleaned_WE_1_DNAseq_YPD_2.fq.gz -3 -l 40 -t 8
El parametro -3 sirve para cortar las bases desde el extremo 3’ que tengan un phred menor a 20 El parametro -l 40 sirve para filtrar reads que tengan un tamaño menor a 40 bp (que pueden existir luego de cortarlas con el parametro -3) El parametro -t 8 indica que usaremos 8 CPUs
Noten que generamos nuevos archivos de reads (con el nombre “cleaned_” al principio). Podemos luego verificar la limpieza de reads corriendo nuevamente FASTQC en los archivos “cleaned_”.
Para continuar con los siguientes modulos, es necesario limpiar todas las reads (usando los mismos parametros que usamos para las reads de DNAseq), y revisar con FASTQC (en conjunto con MULTIQC) la calidad de las reads limpiadas
Key Points
Alineando lecturas al genoma
Overview
Teaching: 60 min
Exercises: 30 minQuestions
¿Como alineo lecturas al genoma?
Objectives
Crear indeces del genoma para los programas de alineamiento
Alinear lecturas de DNAseq y ATACseq al genoma con BWA
Alinear lecturas de RNAseq a los genes anotados en el genoma
Mapeo de reads al genoma y transcriptoma
Nuestras reads son de distintos ensayos: DNAseq, ATACseq, y RNAseq. Una diferencia fundamental entre estos ensayos es el material biológico de donde provienen, que es ADN genómico (DNAseq y ATACseq) o ARN mensajero (RNAseq). Como bien sabemos la estructura del ARNm puede ser distinto al de su templado en el ADN genómico, en especial por la presencia de intrones en el ADN, y esta diferencia es importante en el momento de mapear reads a genomas de referencia. Por esto existen programas especializados en el mapeo de reads genomicas (como BWA), y programas para el mapeo de reads de transcriptoma (RNA STAR).
BWA y RNA STAR requiren realizar un index del genoma de referencia (que les permitirá realizar su trabajo de manera más eficiente), el cual lo tenemos en formato FASTA en la carpeta reference_genome
bwa index reference_genome/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa
[bwa_index] Pack FASTA... 0.23 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 8.41 seconds elapse.
[bwa_index] Update BWT... 0.16 sec
[bwa_index] Pack forward-only FASTA... 0.11 sec
[bwa_index] Construct SA from BWT and Occ... 3.64 sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa index reference_genome/sacCer3.fa
[main] Real time: 14.551 sec; CPU: 12.562 sec
BWA INDEX generó archivos en la carpeta reference_genome Ahora realizamos el index para analizar transcriptomas con RNA STAR. En este caso, también es necesario proveer de la anotación del genoma (en que posicion estan los genes codificantes) que esta en formato GTF.
mkdir star_saccer
STAR --runMode genomeGenerate --genomeDir star_saccer/ --genomeFastaFiles reference_genome/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa --sjdbGTFfile reference_genome/Saccharomyces_cerevisiae.R64-1-1.57.gff3
En este caso fue necesario primero crear una carpeta (star_saccer), donde se guardaron los archivos del index.
Ahora estamos listos para mapear nuestras reads (limpiadas) a los index de BWA y STAR. Primero creamos un directorio donde dejar los archivos de mapeo (bam), y luego ejecutamos BWA para mapear las reads de DNAseq y ATACseq
mkdir bam
bwa mem -t 8 -o bam/WE_1_DNAseq_YPD_1.sam reference_genome/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa DNAseq/cleaned_WE_1_DNAseq_YPD_1.fq.gz DNAseq/cleaned_WE_1_DNAseq_YPD_2.fq.gz
bwa mem -t 8 -o bam/WExNA_1_ATACseq_HIGH_N_1.sam reference_genome/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa ATACseq/cleaned_WExNA_1_ATACseq_HIGH_N_1.fq.gz ATACseq/cleaned_WExNA_1_ATACseq_HIGH_N_2.fq.gz
Luego ejecutamos STAR para cada pareja de reads pareadas
STAR --runThreadN 8 --readFilesIn RNAseq/cleaned_WExNA_1_RNAseq_HIGH_N_1.fq.gz RNAseq/cleaned_WExNA_1_RNAseq_HIGH_N_2.fq.gz --genomeDir star_saccer/ --outFileNamePrefix bam/WENA_1_RNAseq_HIGH_N_ --outSAMtype BAM Unsorted --readFilesCommand zcat --outTmpDir ~/tmp/
STAR --runThreadN 8 --readFilesIn RNAseq/cleaned_WExNA_2_RNAseq_HIGH_N_1.fq.gz RNAseq/cleaned_WExNA_2_RNAseq_HIGH_N_2.fq.gz --genomeDir star_saccer/ --outFileNamePrefix bam/WENA_2_RNAseq_HIGH_N_ --outSAMtype BAM Unsorted --readFilesCommand zcat --outTmpDir ~/tmp/
STAR --runThreadN 8 --readFilesIn RNAseq/cleaned_WExNA_3_RNAseq_HIGH_N_1.fq.gz RNAseq/cleaned_WExNA_3_RNAseq_HIGH_N_2.fq.gz --genomeDir star_saccer/ --outFileNamePrefix bam/WENA_3_RNAseq_HIGH_N_ --outSAMtype BAM Unsorted --readFilesCommand zcat --outTmpDir ~/tmp/
STAR --runThreadN 8 --readFilesIn RNAseq/cleaned_WExNA_1_RNAseq_LOW_N_1.fq.gz RNAseq/cleaned_WExNA_1_RNAseq_LOW_N_2.fq.gz --genomeDir star_saccer/ --outFileNamePrefix bam/WENA_1_RNAseq_LOW_N_ --outSAMtype BAM Unsorted --readFilesCommand zcat --outTmpDir ~/tmp/
STAR --runThreadN 8 --readFilesIn RNAseq/cleaned_WExNA_2_RNAseq_LOW_N_1.fq.gz RNAseq/cleaned_WExNA_2_RNAseq_LOW_N_2.fq.gz --genomeDir star_saccer/ --outFileNamePrefix bam/WENA_2_RNAseq_LOW_N_ --outSAMtype BAM Unsorted --readFilesCommand zcat --outTmpDir ~/tmp/
STAR --runThreadN 8 --readFilesIn RNAseq/cleaned_WExNA_3_RNAseq_LOW_N_1.fq.gz RNAseq/cleaned_WExNA_3_RNAseq_LOW_N_2.fq.gz --genomeDir star_saccer/ --outFileNamePrefix bam/WENA_3_RNAseq_LOW_N_ --outSAMtype BAM Unsorted --readFilesCommand zcat --outTmpDir ~/tmp/
Hemos generado archivos SAM y BAM, que son archivos de alineamiento de reads. Muchos programas que usaremos mas adelante ocupan estos archivos, por ejemplo para visualizar mapeos, encontrar SNPs, contar reads que mapean a ciertos genes, etc. Los archivos SAM y BAM se pueden optimizar para las siguientes tareas, primero ordenandolos y luego haciendo un index. Esto se realiza con samtools
mkdir bam/sorted_bam
samtools sort -O bam -o bam/sorted_bam/WE_1_DNAseq_YPD_1.sorted.bam bam/WE_1_DNAseq_YPD_1.sam
samtools sort -O bam -o bam/sorted_bam/WExNA_1_ATACseq_HIGH_N_1.sorted.bam bam/WExNA_1_ATACseq_HIGH_N_1.sam
samtools sort -O bam -o bam/sorted_bam/WENA_1_RNAseq_HIGH_N_Aligned.out.sorted.bam bam/WENA_1_RNAseq_HIGH_N_Aligned.out.bam
samtools sort -O bam -o bam/sorted_bam/WENA_2_RNAseq_HIGH_N_Aligned.out.sorted.bam bam/WENA_2_RNAseq_HIGH_N_Aligned.out.bam
samtools sort -O bam -o bam/sorted_bam/WENA_3_RNAseq_HIGH_N_Aligned.out.sorted.bam bam/WENA_3_RNAseq_HIGH_N_Aligned.out.bam
samtools sort -O bam -o bam/sorted_bam/WENA_1_RNAseq_LOW_N_Aligned.out.sorted.bam bam/WENA_1_RNAseq_LOW_N_Aligned.out.bam
samtools sort -O bam -o bam/sorted_bam/WENA_2_RNAseq_LOW_N_ligned.out.sorted.bam bam/WENA_2_RNAseq_LOW_N_Aligned.out.bam
samtools sort -O bam -o bam/sorted_bam/WENA_3_RNAseq_LOW_N_Aligned.out.sorted.bam bam/WENA_3_RNAseq_LOW_N_Aligned.out.bam
Ahora aprovechando que todos los archivos que generamos terminan en .sorted.bam, podemos correr samtools index usando un for loop:
for FILE in bam/sorted_bam/*.sorted.bam;do samtools index $FILE;done
Estos archivos nos servirán en la próxima sección para visualizar y realizar informes de calidad de mapeo.
Finalmente, utilizaremos featureCounts para contar la cantidad de lecturas que alinean en ciertas regiones (features) del genoma, en este caso estas regiones serán “genes” segun el archivo GFF3.
mamba install -c bioconda subread
featureCounts -p -a reference_genome/Saccharomyces_cerevisiae.R64-1-1.57.gff3 -t gene -T 8 -o counts.tsv bam/sorted_bam/WENA_1_RNAseq_HIGH_N_Aligned.out.sorted.bam bam/sorted_bam/WENA_2_RNAseq_HIGH_N_Aligned.out.sorted.bam bam/sorted_bam/WENA_3_RNAseq_HIGH_N_Aligned.out.sorted.bam bam/sorted_bam/WENA_1_RNAseq_LOW_N_Aligned.out.sorted.bam bam/sorted_bam/WENA_2_RNAseq_LOW_N_ligned.out.sorted.bam bam/sorted_bam/WENA_3_RNAseq_LOW_N_Aligned.out.sorted.bam
Key Points
Visualizar mapeos y control de calidad
Overview
Teaching: 60 min
Exercises: 30 minQuestions
¿Cómo visualizo las lecturas alineadas al genoma de referencia?
Objectives
Visualización de mapeos con JBROWSE2
JBROWSE nos permite visualizar los archivos bam (que hayan sido ordenados con sorted e indexados con index) Primero debemos cargar el genoma usando OPEN SEQUENCE FILE En Type elegir FastaAdapter, luego Choose file para seleccionar el archivo Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa que esta en la carpeta reference_genome/ Poner un nombre provisorio al genoma en Assembly Name (ejemplo “Saccer3”)
Ahora que tenemos el genoma cargado, podemos abrir los archivos .sorted.bam. También para visualizar la posición de los genes anotados en el genoma, abriremos el archivo .gff3 que esta en la carpeta referece_genome Para una rápida comparación de los tres tipos de ensayos que tenemos, cargamos un archivo sorted.bam de DNAseq, ATACseq, y un archivo de RNAseq:
Para visualizar los mapeos tendremos que acercarnos a una región del genoma
Como pueden ver, los mapeos de reads caen en distintos lugares dependiendo de nuestros ensayos. Mapeos de DNAseq cubren en general todo el genoma, los de ATACseq se concentran en las regiones promotoras, y RNAseq se concentran en las regiones codificantes. También podemos visualizar las variantes que corresponden a las diferencias de nuestras cepas con el genoma de referencia que proviene de la cepa de laboratorio S288C
Analisis de calidad con QUALIMAP
Instalaremos qualimap usando mamba
mamba install -c bioconda qualimap
Generaremos informes HTML de cada archivo de mapeo (sorted.bam) con el siguiente script:
for FILE in bam/sorted_bam/*.sorted.bam;do qualimap bamqc -bam $FILE;done
También usaremos el modulo rnaseq de qualimap para un analisis más detallado de los mapeos de RNAseq
for FILE in bam/sorted_bam/*RNAseq*.sorted.bam;do qualimap rnaseq -bam $FILE -gtf reference_genome/Saccharomyces_cerevisiae.R64-1-1.57.gff3;done
Podemos realizar nuevamente un informe conjunto con MULTIQC
multiqc -o bam/sorted_bam/ bam/sorted_bam/
Key Points
Utilizar herramientas visuales para examinar el alineamiento de lecturas